mTOR inhibitors
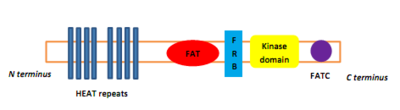
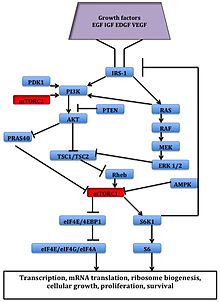
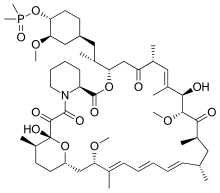
[2][3] Rapamycin was first discovered in 1975 in a soil sample from Easter Island of South Pacific, also known as Rapa Nui, from where its name is derived.[8] The FAT domain consists of repeats, referred to as HEAT (Huntingtin, Elongation factor 3, A subunit of protein phosphatase 2A and TOR1).Several downstream pathways that regulate cell-cycle progression,[12] translation, initiation, transcriptional stress responses,[13] protein stability, and survival of cells are signaling through mTOR.[15] mTORC1 consists of mTOR and two positive regulatory subunits, raptor and mammalian LST8 (mLST8), and two negative regulators, proline-rich AKT substrate 40 (PRAS40) and DEPTOR.In many cancers the PTEN expression is decreased and may be downregulated through several mechanisms, including mutations, loss of heterozygosity, methylation, and protein instability.Due to partial mTOR inhibition as mentioned before, rapalogs are not sufficient for achieving a broad and robust anticancer effect, at least when used as monotherapy.[5] The bacterial natural product rapamycin or sirolimus,[6] a cytostatic agent, has been used in combination therapy with corticosteroids and cyclosporine in patients who received kidney transplantation to prevent organ rejection both in the US[25] and Europe,[26] due to its unsatisfying pharmacokinetic properties.[27] Recently rapamycin has shown effective in the inhibition of growth of several human cancers and murine cell lines.It is approved by the U.S. Food and Drug Administration (FDA)[25] and the European Medicines Agency (EMA),[28] for the treatment of renal cell carcinoma (RCC).[6] Temsirolimus has also been used in a Phase I clinical trial in conjunction with neratinib, a small-molecule irreversible pan-HER tyrosine kinase inhibitor.[30] mTORC1 inhibition by everolimus has been shown to normalize tumor blood vessels, to increase tumor-infiltrating lymphocytes, and to improve adoptive cell transfer therapy.[32] In July and August 2012, two new indications were approved, for advanced hormone receptor-positive, HER2-negative breast cancer in combination with exemestane, and pediatric and adult patients with SEGA.[15] The inhibition of the PI3K/mTOR pathway has been shown to potently block proliferation by inducing G1 arrest in different tumor cell lines.[5] It inhibits T-cell proliferation and proliferative responses induced by several cytokines, including interleukin 1 (IL-1), IL-2, IL-3, IL-4, IL-6, IGF, PDGF, and colony-stimulating factors (CSFs).One of the major stimuli of angiogenesis is hypoxia, resulting in activation of hypoxia-inducible transcription factors (HIFs) and expression of ANG2, bFGF, PDGF, VEGF, and VEGFR.[2][38] Pharmacologic down-regulation of (mTOR) pathway during chemotherapy in a mouse model prevents activation of primordial follicles, preserves ovarian function, and maintains normal fertility using clinically available inhibitors INK and RAD.These mTOR inhibitors, when administered as pretreatment or co-treatment with standard gonadotoxic chemotherapy, helps to maintain ovarian follicles in their primordial state.[44] Reduction of the inflammatory cytokine Interleukin 1 beta (IL-1β) in mice by mTOR inhibition (with rapamycin in doses of 20 mg/kg/day, human equivalent about 1.6 mg/kg/day[43]) has been shown to enhance learning and memory.The structural characteristics common to temsirolimus and sirolimus; the pipecolic acid, tricarbonyl region from C13-C15, and lactone functionalities play the key role in binding groups with the FKBP12.[2] Nonetheless, the rapamycin analogs that have been approved for human use are modified at C-43 hydroxyl group and show improvement in pharmacokinetic parameters as well as drug properties, for example, solubility.The most frequently occurring adverse events are stomatitis, rash, anemia, fatigue, hyperglycemia/hypertriglyceridemia, decreased appetite, nausea, and diarrhea.mTORi-induced ILD often is asymptomatic (with ground glass abnormalities on chest CT) or mild symptomatic (with a non-productive cough), but can be very severe as well.[49] Identification of predictive biomarkers of efficacy for tumor types that are sensitive to mTOR inhibitors remains a major issue.In a recent study of patients with Renal cell carcinoma, resistance to Temsirolimus was associated with low levels of p-AKT and p-S6K1, that play the key role in mTOR activation.For future studies, it is recommended to exclude patients with low or negative p-AKT levels from trials with mTOR inhibitors.[20] Several, so-called mTOR/PI3K dual inhibitors (TPdIs), have been developed and are in early-stage preclinical trials and show promising results.Studies have shown superior antiproliferative activity to rapalogs and in vivo models have confirmed these potent antineoplastic effects of dual mTOR/PI3K inhibitors.[1][7] These inhibitors target isoforms of PI3K (p110α, β and γ) along with ATP-binding sites of mTORC1 and mTORC2 by blocking PI3K/AKT signaling, even in cancer types with mutations in this pathway.Tumors that depend on PI3K/mTOR pathway should respond to these agents but it is unclear if compounds are effective in cancers with distinct genetic lesions.[7] A way to overcome the resistance and improve efficacy of mTOR targeting agents may be with stratification of patients and selection of drug combination therapies.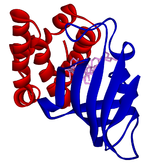



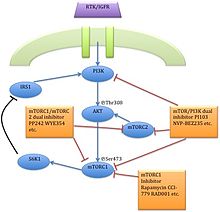
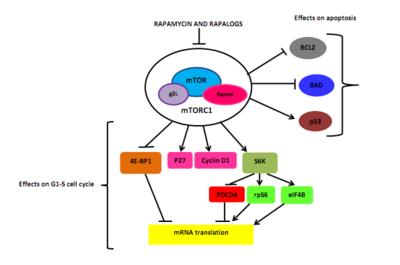
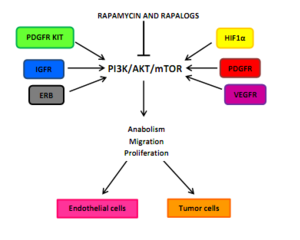

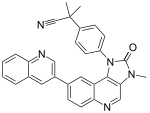
Drug classImmunosuppressionMechanism of actionBiological targetFKBP12class of drugsmammalian target of rapamycinserine/threonine-specific protein kinasephosphatidylinositol-3 kinaseprotein complexesmTORC1mTORC2inhibitorrapamycinEaster IslandSouth PacificmacrolidemicroorganismStreptomyces hygroscopicusantifungalimmunosuppressivecellular proliferationcell cycle progressionRibbon diagramprotein kinasesprotein tyrosine kinasesprotein serine/threonine kinasesDual-specificity kinasesphosphatidylinositol-3 kinase-related kinases (PIKKs)N-terminusC-terminusthe kinase domainα-helicesHuntingtinElongation factor 3protein phosphatase 2Asmall moleculebiologicsphosphorylationsubstratesautophosphorylationgrowth factorsamino acidsoxygendownstream pathwaystranslationinitiationproteinsurvival of cellsserine/threonine kinasePI3K/AKTmultiprotein complexesraptorrictorphosphorylatespaxillinGTPasemigrationactin cytoskeletonmRNA translationamplificationmutationtherapeutic targetgrowth factor receptorsTumor suppressor phosphatase and tensin homologuechromosome 10mutationsloss of heterozygositymethylationeukaryotic initiation factor 4E-binding protein 1 (4EBP1)cellular transformationCandida albicansAspergillus fumigatusCryptococcus neoformanstransplant rejectioncyclosporine Arenal transplantationtoxicitycytostaticre-stenosisneurodegenerative diseasestemsirolimuspharmacokineticstreatment of cancereverolimusridaforolimusclinical trialshydrophilicityintravenous administrationNational Cancer Institutemonotherapycombination therapypreclinicalnegative feedbacksmall molecule inhibitorsnatural productsirolimuscytostatic agentcorticosteroidscyclosporinekidney transplantationorgan rejectionU.S. Food and Drug Administrationcoronary arteriesatherosclerosisprodrugFood and Drug AdministrationEuropean Medicines Agencyneratinibtyrosine kinase inhibitornauseastomatitisanemiatumor-infiltrating lymphocytesadoptive cell transfer therapysunitinibsorafenibsubependymal giant cell astrocytomatuberous sclerosissarcomaUmirolimusZotarolimuscoronarytorin-1catalyticcell cycle