Rosetta@home
Rosetta@home aims to predict protein–protein docking and design new proteins with the help of about fifty-five thousand active volunteered computers processing at over 487,946 GigaFLOPS on average as of September 19, 2020.[6] With an influx of new users looking to participate in the fight against the COVID-19 pandemic, caused by SARS-CoV-2, Rosetta@home increased its computing power up to 1.7 PetaFlops as of March 28, 2020.A primary feature of the Rosetta@home graphical user interface (GUI) is a screensaver which shows a current workunit's progress during the simulated protein folding process.In the upper-left of the current screensaver, the target protein is shown adopting different shapes (conformations) in its search for the lowest energy structure.[26] One of the main goals of Rosetta@home is to predict protein structures with the same accuracy as existing methods, but in a way that requires significantly less time and money.Rosetta@home also develops methods to determine the structure and docking of membrane proteins (e.g., G protein–coupled receptors (GPCRs)),[27] which are exceptionally difficult to analyze with traditional techniques like X-ray crystallography and NMR spectroscopy, yet represent the majority of targets for modern drugs.High scoring groups in this sometimes competitive experiment are considered the de facto standard-bearers for what is the state of the art in protein structure prediction.In the 2004 CASP6 experiment, Rosetta made history by being the first to produce a close to atomic-level resolution, ab initio protein structure prediction in its submitted model for CASP target T0281.[35] This was inspired in part by the retraction of a high-profile paper from 2004 which originally described the computational design of a protein with improved enzymatic activity relative to its natural form.[41] A component of the Rosetta software suite, RosettaDesign, was used to accurately predict which regions of amyloidogenic proteins were most likely to make amyloid-like fibrils.[43] Rosetta@home was used in the same study to predict structures for amyloid beta, a fibril-forming protein that has been postulated to cause Alzheimer's disease.The computer model accurately predicted docking between LF and PA, helping to establish which domains of the respective proteins are involved in the LF–PA complex.[51] As part of research funded by a $19.4 million grant by the Bill & Melinda Gates Foundation,[52] Rosetta@home has been used in designing multiple possible vaccines for human immunodeficiency virus (HIV).[53][54] In research involved with the Grand Challenges in Global Health initiative,[55] Rosetta has been used to computationally design novel homing endonuclease proteins, which could eradicate Anopheles gambiae or otherwise render the mosquito unable to transmit malaria.[56] Being able to model and alter protein–DNA interactions specifically, like those of homing endonucleases, gives computational protein design methods like Rosetta an important role in gene therapy (which includes possible cancer treatments).[38][57] In 2020, the Rosetta molecular modelling suite was used to accurately predict the atomic-scale structure of the SARS-CoV-2 spike protein weeks before it could be measured in the lab.The small size and high stability of the inhibitors is expected to make them adequate to a gel formulation that can be nasally applied or as a powder to be administered directly onto the respiratory system.[72] Originally introduced by the Baker laboratory at the University of Washington in 1998 as an ab initio approach to structure prediction, Rosetta has since branched into several development streams and distinct services, providing features such as macromolecular docking and protein design.[76] Rosetta and RosettaDesign earned widespread recognition by being the first to design and accurately predict the structure of a novel protein of such length, as reflected by the 2002 paper describing the dual approach prompting two positive letters in the journal Science,[77][78] and being cited by more than 240 other scientific articles.[87] The vastly increased computing power afforded by the Rosetta@home network, combined with revised fold-tree representations for backbone flexibility and loop modeling, made RosettaDock sixth out of 63 prediction groups in the third CAPRI assessment.[6][34] The Robetta (Rosetta Beta) server is an automated protein structure prediction service offered by the Baker laboratory for non-commercial ab initio and comparative modeling.Robetta tasks run on Baker lab servers, Janelia Research Campus machines, and Rosetta@home participant computers.For domains that have no detected structural homologs, a de novo protocol is followed in which the lowest energy model from a set of generated decoys is selected as the final prediction.[97] The game gives users a set of controls (for example, shake, wiggle, rebuild) to manipulate the backbone and amino acid side chains of the target protein into more energetically favorable conformations.[112][113] Although he now uses it to create databases for biologists, Richard Bonneau, head scientist of the Human Proteome Folding Project, was active in the original development of Rosetta at David Baker's laboratory while obtaining his PhD.As of March 28, 2020[update], about 53,000 users from 150 countries were active members of Rosetta@home, together contributing idle processor time from about 54,800 computers for a combined average performance of over 1.7 PetaFLOPS.[122] Rosetta@home users who predict protein structures submitted for the CASP experiment are acknowledged in scientific publications regarding their results.[33] Users who predict the lowest energy structure for a given workunit are featured on the Rosetta@home homepage as Predictor of the Day, along with any team of which they are a member.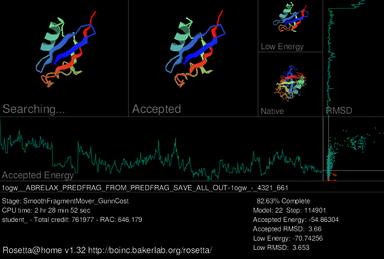
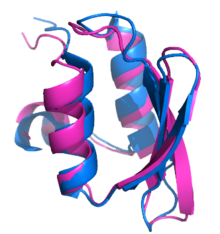


Developer(s)Stable releaseOperating systemWindowsAndroidPlatformLicenseProprietaryfreewarevolunteer computingprotein structure predictionBerkeley Open Infrastructure for Network Computingprotein–protein dockingdesign new proteinsFolditcrowdsourcingbasic researchproteomicsapplied researchmalariaAlzheimer's diseaseworkunitsserverdatabasescross-platformproteinstructural bioinformaticsHuman Proteome Folding ProjectCritical Assessment of Techniques for Protein Structure PredictionCritical Assessment of Prediction of Interactionstertiary structureCOVID-19 pandemicSARS-CoV-2IVX-411clinical trialGBP510NL-201List of volunteer computing projectscentral processing unitclock speedmegabytesdisk spacephysical memoryHypertext Transfer ProtocolfirewallUniversity of Washingtonrandom seedenergy landscapeglobal minimumnative statescreensaverubiquitingraphical user interfaceworkunitprotein foldingthermodynamic free energyroot-mean-square deviationProtein dockingProtein designgenome sequencing projectsprimary structuresuperpositionedÅngströmX-ray crystallographynuclear magnetic resonanceNational Center for Biotechnology InformationProtein Data Bankmembrane proteinsG protein–coupled receptorsstructural homologysequence homologyconformation spaceLevinthal's paradoxincrease exponentiallycomplexed proteinsquaternary structureprotein interactiondrug designresearch paperproof of conceptgreen chemistrybioremediationNobel Prize in Chemistryamyloidamyloid betaanthrax toxindomainsantibodyimmunoglobulin Gherpes simplex virus 1Bill & Melinda Gates FoundationGrand Challenges in Global HealthAnopheles gambiaegene therapySciencemonoclonal antibodiesSK BioscienceIL-2 receptorNew YorkerInvestigational New DrugFood and Drug Administrationsource-availableBaker laboratorymacromolecular dockingRosetta StoneFortranobject-orientedProtein GsuperpositionDavid Baker'sUniversity of North Carolina, Chapel Hillalgorithmporcineα-amylasecamelidJohns Hopkins Universityloop modelingCAMEO3DPSI-BLAST3D-JuryPfam databasehomology modelinglowest energyMonte Carloprotein contact mapside chains